The software system provides an electronic questionnaire containing the user's habits, eating habits, physiological indicators, sickness, occupational history, family history, personal physical exercise, environmental status and other information for the user to fill. After the information is submitted, the system gives the health risk assessment report of the tumor, cardiovascular and cerebrovascular diseases according to the user's personal information (gender, age) and the submitted questionnaire. After extraction of information from various diseases (tumor: lung cancer, liver cancer, colon cancer, stomach cancer, esophageal cancer, bladder cancer, cardiovascular and cerebrovascular: coronary heart disease, stroke, diabetes) in the system, disease-related risk factors were derived. The disease assessment report was derived by risk factors rating score table. The optimal improvement measures will be given according to the relevant knowledgebase.
BioIE is a DEMO of biomedical named entity (drug, disease, gene, protein, etc.) annotation and their relation extraction from biomedical texts. The Demo is implemented with Deep learning and Natural Language Processing techniques. A knowledge graph of Hepatocellular carcinoma (HCC) is also presented.
The Precision Medicine Ontology (PMO) has been developed as a standardized ontology for human precision medicine with consistent, reusable and sustainable descriptions of human disease terms, genomic molecular, phenotype characteristics and related medical vocabulary disease concepts through collaborative efforts of researchers at Institute of Medical Information, Chinese Academy of Medical Sciences. The Precision Medicine Ontology focuses on the development of a high level patients-centric ontology for the academic research, thus, it systematically integrates biomedicine vocabularies from GO, NCBI Gene, DrugBank, HPO, MeSH, SNOMED CT, etc.
Precision Medicine Corpus (PMCorpus) is a public resource providing a manually annotated corpus and related resources for information extraction in the biomedical domain. This is the first golden standard corpus in biomedical field in China. Based on the Precision Medicine Ontology, PMCorpus contains 6000 PubMed articles in six major fields, including cancer, cardiovascular and metabolic diseases. This corpus is an essential resource for biomedical text mining, and is the basic support for semantic annotation, machine translation, knowledge correlation, data mining, intelligent retrieval and other functions.
As large amounts of data accumulate in the field of precision medicine and the semantic relationships between biomedical entities become more and more complex.The construction of precision medicine ontology needs a lot of collaboration of experts in many areas.The collaborative working platform we built for precision medicine ontology construction is an interactive online application for collaboratively editing, browsing, and sharing of precision medicine ontology.The collaborative working platform guarantees the comprehensiveness and instantaneity in the process of ontology construction.
BATMAN-TCM is the first online bioinformatics analysis tool specially designed for the research of molecular mechanism of TCM, mainly based on TCM ingredients’ target prediction and the following network pharmacology analyses of the potential targets, aiming to contribute to the understanding of the “multi-component, multi-target and multi-pathway” combinational therapeutic mechanism of TCM and to provide clues for the following experimental validation.
The Chromosome-centric Human Proteome Project (C-HPP) aims to catalog the genome-encoded proteins on the chromosome-by-chromosome basis. As the C-HPP proceeds, the increasing requirement of data-intensive analyses for the MS/MS data poses a challenge to the proteomic community, especially those small laboratories lacking computational infrastructures.
To address this challenge, we update the previous CAPER browser into a higher version, CAPER 3.0 – a scalable cloud-based system for the data-intensive analysis of C-HPP datasets. CAPER 3.0 uses cloud computing technology to facilitate MS/MS-based peptide identification. In particular, it can use both public and private cloud, aiming to help analyze the C-HPP datasets.
Self-interacting proteins, whose two or more copies can interact with each other, play important roles in cellular functions and the evolution of protein interaction networks. Knowing whether a protein can self-interact can contribute to and sometimes is crucial for the elucidation of its functions. SLIPPER (SeLf-Interacting Protein PrEdictoR) is designed to predict whether a protein can interact with itself, and meanwhile provide various related annotation information to facilitate its further functional research.
UbiBrowser is a resource of known and predicted human ubiquitin ligase (E3) - substrate interaction network.
The E3-substrate interactions are derived from severn data sources: mannual cruration, protein ortholog, protein domain, protein motif and network topology.
A computational framework is used to to combine multiple biological evidences to generate a confidence score using Naïve Bayesian Network.
High-throughput screens inevitably generate a significant number of false positives, which result in erroneous data and thus misleading our conclusions. Strategies should be taken to assess the confidence of high-throughput data and minimize the high degree "false-positive". Here we introduce the Bayesian method to evaluate the potential biological relevance of the identified interactions.
Autoantibodies are antibodies that are produced by the B-cell immune system against an individual’s own antigens and play pivotal roles in the maintenance of healthy individuals’ homeostasis, as well as in tumors and autoimmune diseases. In the last two decades, tremendous efforts have been devoted to elucidate the generation, evolution and function of autoantibodies and their targets, autoantigens [1-9]. However, the previously identified autoantigens are randomly dispersed in the literature.
PNmerger (biological Pathway and protein Netwrok merge) is a java based plug-in for the widely used open-source Cytoscape molecular interaction viewer. For an interaction network, PNmerger will automatically annotate the network proteins with the KEGG pathway information, to find the known pathway elements in protein network, and also to predict the possible pathway elements. To present the pathway information for the protein network , PNmerger can illustrate the clusters of the nodes with the same biological pathway, and also present the the potential crosstalks between different pathways. This information will be helpful for the users to find the important clues for knowledge discovery and also experimental design.
UsIng MedPortal to access and share ontologies. You can create ontology-based annotations for your own text , link your own project that uses ontologies to the description of those ontologies , find and create relations between terms in different ontologies, review and comment on ontologies and their components as you browse them. Sign in to MedPortal to submit a new ontology or ontology-based project, provide comments on ontologies or add ontology mappings.
Data analysis poses a significant challenge to the large-scale proteomics studies. Based on the structured and controlled vocabularies - Gene Ontology (GO), and the GO annotation from related databases, a strategy (named GOfact) is developed to identify the functional distribution and the significantly enriched functional categories of the proteomic expression profile. It would be helpful for understanding the overall functions of these identified proteins and supply the fundamental information for further bioinformatics exploration.
HisgAtlas is a manual curation database for human immunosuppression genes. Immunosuppression is body’s state in which the activation or efficacy of immune system is weakened. It is associated with a wide spectrum of human diseases, such as autoimmune diseases, allergy, organ transplantation rejection and chronic infectious diseases. In the last two decades, tremendous efforts have been made to elucidate the mechanism of hundreds of immunosuppression genes. For example, The programmed cell death-1 (PD-1) receptor, a cell surface receptor on T cells,which plays a critical role in peripheral tolerance, can also compromise antiviral and antitumor T cell responses through the inhibition of T cell proliferation. Blockade of the PD-1/PD-L1 pathway can active antitumor immune responses and have been a very successful therapy for cancer. Immunosuppression genes could be valuable drug targets or biomarkers for the immunotherapeutic treatment of different diseases. However, the information of all previously identified immunosuppression genes is dispersed in thousands of publications. Here we provided the HisgAtlas database that collects 995 previously identified human immunosuppression genes using text mining and manual curation.
OsteoporosAtlas is a manually curated database for human osteoporosis-related genes. Osteoporosis is a common disorder with a strong genetic component characterized by reduced bone mass and increased risk of fragility fractures. Hundreds of osteoporosis-related genes have been identified in thousands of publications, which involve in the pathogenesis of individual cases, novel diagnostic and prognostic biomarker, individual treatment responses and precision medicine. Here we present a database containing 617 osteoporosis-related encoding genes, 131 microRNAs, determined by text-mining and manual curation.
UVGD is a manual curation database for ultraviolet-related genes. Exposing to ultraviolet for a certain time always triggers some significant molecular effects in organisms. Hundreds of genes have been identified to be responsible for the molecular biology effect of ultraviolet radiation. In the current version of UVGD, there are 663 ultraviolet-related genes collected by literature-mining and manual curation.
PIC(paediatric Intensive Care) database is a large, single-center database comprising information relating to patients admitted to critical care units at the Children's Hospital of Zhejiang University School of Medicine.
A pediatric disease map shows the relationships among the most common pediatric diseases, incidence rates, ages of onset and seasonal patterns in children based on more than 5 million outpatient visits at The Children's Hospital Zhejiang University School of Medicine (ZUCH).
RDmap is the first user-interactive map-style rare disease knowledgebase. It will help clinicians and researchers explore the increasingly complicated realm of rare genetic diseases.Total 3287 rare diseases are included in the phenotype-based map, and 3789 rare genetic diseases are included in the gene-based map; 1718 overlapping diseases are connected between two maps.
This is a DEMO of adverse drug reaction search engine (ADRSE) which takes a drug name as input and listed related ADRs and their references. The Demo is implemented with Deep Learning and Natural Language Processing techniques. Specifically, over 26,000 Pubmed abstracts (1975-2020) are retrieved and then, drug mentions are recognized by utilizing BiLSTM-CRF network and ADR mentions are recognized by using BiLSTM-GAT-CRF network. The relations between drugs and ADRs are extracted using both sentence- and document-level relation extraction methods (See the references for the specific methods). Finally, all the relations are integrated.
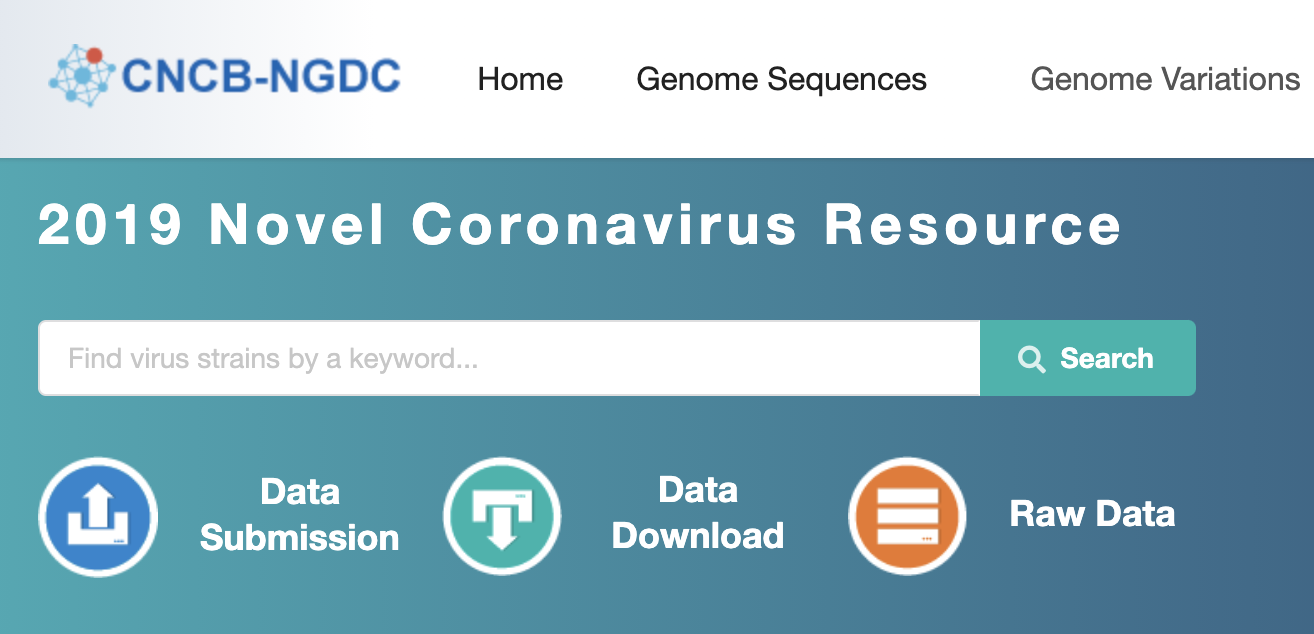
2019nCoVR features comprehensive integration of genomic and proteomic sequences as well as their metadata information from the GISAID, NCBI, NMDC and CNCB/NGDC. It also incorporates a wide range of relevant information including scientific literatures, news, and popular articles for science dissemination, and provides visualization functionalities for genome variation analysis results based on all collected SARS-CoV-2 strains.
The BioSample database provides structured and indexed descriptive information on biological samples. BioSample also provides reciprocal links to BioProject as well as other relevant database resources, facilitating sample search in different databases. BioSample features batch submission and accepts diverse data types including human, plant, animal, microbe, virus, metagenome, etc.
The BioProject database serves as an organizational framework to provide a centralized access to descriptive metadata about research projects, ranging from genomic, transcriptomic, epigenomic and metagenomic sequencing efforts to genome-wide association studies and variation analyses. BioProject features umbrella project, providing an organizational structure for any large project.
GWH shortens for Genome Warehouse, a data repository for genome assembly data. It archives genome assembly sequence, genome annotation and other associated data. GWH is one of database resources in BIG Data Center (BIGD), part of Beijing Institute of Genomics (BIG), Chinese Academy of Sciences (CAS), serving as a primary archive of genome assembly associated data for worldwide institutions and laboratories.
The transcriptional factor regulatory network graph combines the results obtained from multiple databases, and is networked and displayed through Cytoscape. The blue stars represent transcription factors, and the red circles represent downstream regulatory genes. The dropdown box is used to screen and control the display of the page.
PPI protein interactions were analyzed and the community structure was obtained by clustering algorithm. By default, a "modular greedy optimization" clustering method is used. The resulting page is presented in two formats, one in tabular form and the other in network graph style.
LncBook is a curated knowledgebase of human lncRNAs that features a comprehensive collection of human lncRNAs and systematic curation of lncRNAs by multi-omics data integration, functional annotation and disease association. The current implementation of LncBook houses a large number of 270,044 lncRNAs and includes 1,867 featured lncRNAs with 3,762 lncRNA-function associations. It also integrates an abundance of multi-omics data from expression, methylation, genome variation and lncRNA-miRNA interaction. In addition, LncBook includes 3,772 experimentally validated lncRNA-disease associations and identifies a total of 97,998 lncRNAs that are putatively disease-associated. To facilitate online analysis, a series of useful tools such as coding potential prediction, sequence search, etc., are deployed in LncBook.
EWAS Atlas is dedicated to the curation, integration and standardization of EWAS knowledge and has the great potential to help researchers dissect molecular mechanisms of epigenetic modifications associated with biological traits.
iDog, an integrated resource for domestic dog (Canis lupus familiaris) and wild canids, provides the worldwide dog research community a variety of data services. This includes Genes, Genomes, SNPs, Breed/Disease Traits, Gene Expressions, GO Function Annotations, Dog-Human Homolog Diseases and Literatures. In addition, iDog provides Online tools for performing genomic data visualization and analyses.
The Genome Variation Map (GVM) is a public data repository of genome variations, including single nucleotide polymorphisms and small insertions and deletions, with particular focuses on human as well as cultivated plants and domesticated animals.
The Methylation Bank (MethBank) is a comprehensive database that features consensus reference methylomes (CRMs) and single-base resolution methylomes (SRMs) across a variety of species and integrates epigenome-wide associations and DNA & RNA methylation tools.
With the rapid development of sequencing technologies towards higher throughput and lower cost, sequence data are generated at an unprecedentedly explosive rate. To provide an efficient and easy-to-use platform for managing huge sequence data, here we present Genome Sequence Archive (GSA; http://bigd.big.ac.cn/gsa or http://gsa.big.ac.cn), a data repository for archiving raw sequence data. In compliance with data standards and structures of the International Nucleotide Sequence Database Collaboration (INSDC), GSA adopts four data objects (BioProject, BioSample, Experiment, and Run) for data organization, accepts raw sequence reads produced by a variety of sequencing platforms, stores both sequence reads and metadata submitted from all over the world, and makes all these data publicly available to worldwide scientific communities. In the era of big data, GSA is not only an important complement to existing INSDC members by alleviating the increasing burdens of handling sequence data deluge, but also takes the significant responsibility for global big data archive and provides free unrestricted access to all publicly available data in support of research activities throughout the world.